



中国药典回收率、精密度相关规定解读
最近在药品研发申报中遇到省药检所老师拒绝受理方法复核,理由为有关物质方法验证不符合2015版《中国药典》9101项下回收率、精密度的相关规定。我们该怎么样应对,本文从:1) 9101关于回收率、精密度相关规定的溯源。2) 2020版《中国药典》两次征求意见对比。3、各国法规指南对该项可接受标准的相关规定。三个方面展开论述,明确9101关于回收率、精密度相关规定的适合使用的范围,科学的进行方法验证,确保药品的质量可控。
通则1200适合使用的范围,1、注重于在USP-NF决定是不是收载与某项测试相关的分析方法及其验收标准前,USP希望看到的数据类型。2、提供了一套系统的验证方法和标准用以评估验证数据。可提供满足本通则要求的验证数据的分析方法适用于药典,也可适用于监管的考量。3、不是为满足公司对应用于生产基地的分析方法的验证的需求。
通则1200对于LOD的新定义,1、配置一个在限度浓度的杂质标准品溶液。2、配置一个添加了在限度浓度的杂质标准品的样品标准品溶液。3、配置一个添加了(限度100%-RSD%*浓度)的杂质标准品的样品标准品溶液。如果12且3<2,则差异可以被检测出,否则,该方法不能胜任限度检测。*从Horwitz方程估算得出。
个人理解,PF39(6)1200淘汰了检测限、定量限,提出了新的“可检测性”,更专注于精密度和准确度的结果。2020版《中国药典》9101分析方法验证指导原则只是对方法的准确度和精密度做出了相关规定,但并未对方法的灵敏度做相应的更改。根据PF39(6)1200相关理念,方法的精密度验证中待测样品应添加规定限度的指定杂质对照品,以加样回收率RSD%来评估方法的测定精度。因此今后的方法验证中应采用加标样品进行方法的精密度验证。定量限也需重新定义为满足准确度及精密度有关要求的最低浓度,样品中的杂质含量若小于定量限应报告为LOQ。
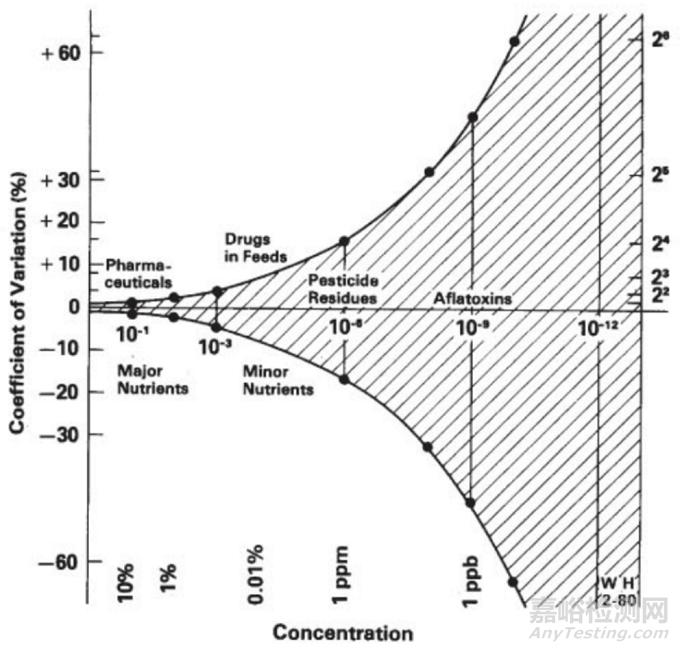
个人理解,该规定可当作收载入药典的方法的有关要求,但在药品研发过程强制实施,可能会事与愿违,USP1220也提出了分析方法的生命周期管理,药物研发处于药品产业链的上游,分析方法只要能够完全满足药品的安全、有效、质量可控的评估即可,关于方法本身的准确度及精密度没有最好,只有更好,是药品全生命周期中逐步完善的一部分。
关于2020版《中国药典》9101项下关于回收率及精密度的多次征求意见稿如下表所示:
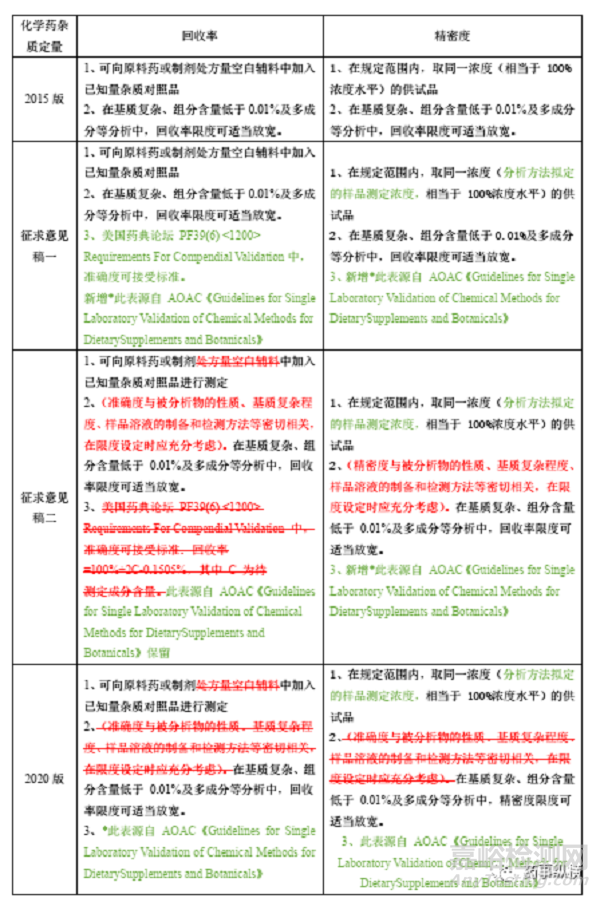
个人理解,由上表可知药典委在进行准确度、精密度的相关修订时肯定也考虑了不少的问题,但有三处疑惑,
1、修订第一稿的时候引入了PF39(6)1200Requirements For Compendial Validation,修订第二稿时又将其删除 ;
2、第二次修订稿中明确(回收率、精密度与被分析物的性质、基质复杂程度、样品溶液的制备和检测的新方法等紧密关联,在限度设定时应最大限度地考虑),但在2020版正式稿中,该句话却没有被收载。
3、在基质复杂、组分含量低于0.01%及多成分等分析中,回收率、精密度可适当放宽,制剂辅料通常种类多、占比重,是否属于限度可适当放宽范畴,有关物质方法通常都是同时测定十几种杂质是否属于多组分分析。
药物研发属于产业链的上游,分析方法处于成型阶段,目前最新的ICHQ2(R1)及USP1225中也并未对重复性及回收率的可接受标准明确规定,但精确指出“接受标准将取决于含量和分析物的性质”。综上所述,该限度要求作为药物研发分析验证的标尺,对于有些品种,可能没办法满足,在审评中应考虑真实的情况,不能一刀切。
1、精密度应该用同一均匀,权威样品进行考察。然而,若无法获得同一均匀样品,可以用人工制备样品或样品溶液进行考察。
至少6次测定(同一溶出杯样品连续进针6次);溶出曲线可只比较最低浓度与最高浓度(USP1092)。
中间精密度的可接受标准或耐用性是预先确定的,溶出度小于 85% 的时间点,RSD不能超过 10.0%;大于 85%的时间点,RSD不能超过 5.0%。
例如在杂质定量测试中,回收率 90 %时一般认为是可接受的。在清洁验证中,如果回收率90 %,在M残留量真值计算中可以不需要仔细考虑回收率;如果回收率90%,则需要在M计算时加以考虑(参见公式 4),如果回收率50%,则该方法不适用。
个人理解,从国内外相关法规指南,对于方法验证中的精密度及回收率的考察的可接受标准不完全一样,但均需满足确保药品安全、有效、质量可控的底线,分析方法的精度与准确度是我们不懈追求的目标,但也需权衡仪器设施、人员及时间成本。
本文从“9101关于回收率、精密度相关规定的溯源”、“2020版《中国药典》两次征求意见对比”、“各国法规指南对该项可接受标准的相关规定”三个方面展开论述,目的是说明:
1、分析方法的准确度和精密度是分析工作者不断追求的目标,如不满足《中国药典》9101项目下的相关规定,需从如何确定保证产品的安全、有效和质量可控角度进行说明。
2、《中国药典》9101项目下精密度、准确度项下的相关规定更适宜作为收载入药典标准的一个验收标准,若想在药物研发过程中实施,需明确适合使用的范围,毕竟“在基质复杂、组分含量低于0.01%及多成分等分析中可适当放宽”这句话在日常工作中不好把控。

【医药答疑】《中国药典》四部的检验测试的项目及检测报告 [2023.09.27]
国内外制药企业质量管理成熟度现状及对我国药品生产监管的启示 [2023.09.18]
药典委:公示药包材及组件11个通则公示和重点说明内容 [2023.09.16]
【药研日报0912】纽安津mRNA肿瘤疫苗启动IIT临床 FIC溃肠炎新药中国获批临床... [2023.09.12]
【药研日报0928】上海甫康PI3K β/δ抑制剂获批Ⅱ期临床 和誉FGFR4抑制剂美国获批肝癌临床...
《医疗器械真实世界研究设计和统计分析注册审查指导原则》公开征求意见(附全文)
腹腔内窥镜手术系统注册审查指导原则第2部分:动物试验决策判定和要求公开征求意见(附全文)
《高频手术设备注册审查指导原则 (2023年修订版)》公开征求意见(附全文)
【药研日报0904】亚虹DBH抑制剂国内获批UC临床 信诺维HPK1激酶抑制剂获批临床...


